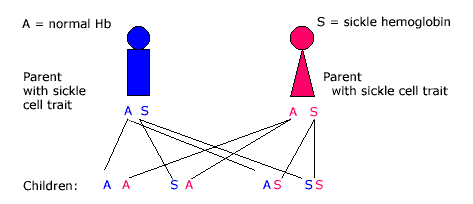
Sickle cell disease requires lifelong comprehensive care. At Children's Wisconsin, our sickle cell care begins at diagnosis. Families often learn their newborn has sickle cell disease from the Wisconsin Newborn Screening Program.
Children's Wisconsin has the state's largest comprehensive pediatric sickle cell disease program. We care for the whole child, from physical health to social and emotional well-being.
Our care team includes:
- Pediatric hematologists: A hematologist with expertise in sickle cell disease care is on call 24 hours a day, 7 days a week for children with sickle cell disease who need urgent medical attention.
- Nurse coordinators: Manage and schedule care for our patients.
- Nurse practitioner: Manages care for our patients.
- Neuropsychometrist: An expert in how sickle cell disease affects the brain. We test and screen patients and work with their schools.
- Psychologist: Supports our kids and families. They offer behavior management, psychological screening and provide ongoing counseling.
- Social worker: Helps our families access the resources and support they need.
- Clinical navigator: A certified Sickle Cell Disease Community Health Worker. They guide our families to resources in the community and work to improve the holistic health of the children cared for in our program.
- Genetic counselor: Helps families understand the genetics of sickle cell disease and what it means for their family. They also discuss what a diagnosis means for eventual family planning.
- Administrative coordinator: Supports our program, including scheduling and communicating with families.
- Researchers: Our physician researchers and clinical research coordinators work to learn more about sickle cell disease and its treatments.
At a patient's first visit, typically by 2 months of age, the child and family meet with our care team. They begin a treatment plan and learn about sickle cell disease and its care. Complications can come on suddenly and without warning. We make sure parents know when and how to seek care.
Patients have follow-up visits at six months and one year. After that, we see them annually or biannually for comprehensive visits.
We see kids on hydroxyurea every two to three months in our outpatient clinic. Kids who often need blood transfusions or other therapies come every month.
Clinic visits include:
- Meetings with the care team
- In-clinic tests
- Medicine refills
- Imaging
- Vaccines
We work closely with the Jane B. Pettit Pain and Headache Center to help manage kids' pain. We also work closely with the emergency department to ensure children with sickle cell disease receive the best and safest care. We work with the pulmonary team to ensure children with sickle cell disease and asthma or other lung complications receive coordinated care.
We manage sickle cell disease care until age 19. Our young adults then transition to seeing adult sickle cell disease experts. We refer local young adults to the Froedtert Hospital's Sickle Cell Disease Clinic. Our sickle cell disease team works with Froedtert to ensure a smooth transition.
Several factors predict the long-term outcomes of sickle cell disease.
They include:
- Type of sickle cell disease
- Severity
- How often and the type of complications that arise
- If your child can stick with their care plan
The life expectancy has increased over the past 30 years. Those with sickle cell disease now regularly live into their mid-40s and beyond. New treatments have improved the prognosis for these kids, but it is still a severe and chronic disease that requires careful and close management with a sickle cell disease expert.